|
AQP genes have
been identified in many land plant species (Johansson et al., 2000). We
conducted a phylogenetic analysis that included a number of AQPs from other
plants to determine how the maize sequences relate to sequences of other
proteins whose properties have already been studied.
Only four of
these proteins have been examined for water channel activity in oocytes.
ZmTIP1-1 (Chaumont et al., 1998) and Zm PIP2-5 (Chaumont et al., 2000) have
high activity, whereas ZmPIP1-1 and ZmPIP1-2 are inactive in the oocyte
assay. The activities of all others remain to be studied.
Phylogenetic Analysis of ZmPIPs
The PIP subfamily
differs from the TIP subfamily by the presence of an additional 20 to 38
amino acids at the N terminus of the PIP proteins. In addition, there are
a large number of amino acid positions (142, or about 50%) that are conserved
in all PIPs. The PIPs can be divided into two major groups, referred to
as PIP1 and PIP2 (Fig. 3), in accordance with the work of Kammerloher et
al. (1994) and others subsequently. All PIP2 proteins examined for water
channel activity in Xenopus laevis oocytes show good activity, but PIP1
proteins are often inactive in oocytes (Chaumont et al., 2000). The reason
for this difference is not known. Some other AQPs such as soybean (Glycine
max) Nod26 and mammalian AQP0 have very low activity in the X. laevis oocyte
assay. PIP2 proteins are characterized by a shorter N-terminal extension
than PIP1 proteins and a longer C-terminal end that contains putative phosphorylation
sites (Schäffner, 1998; Chaumont et al., 2000; Johansson et al., 2000).
In addition to several conservative amino acid substitutions between PIP1s
and PIP2s, some positions show single nonconservative exchange associated
with each subgroup (i.e. Gly 56 Val 44 in TM1, Gln 90 Leu 86 in TM2, and
Met 140 Ala136 in TM3 in ZmPIP1-1 and ZmPIP2-1, respectively).
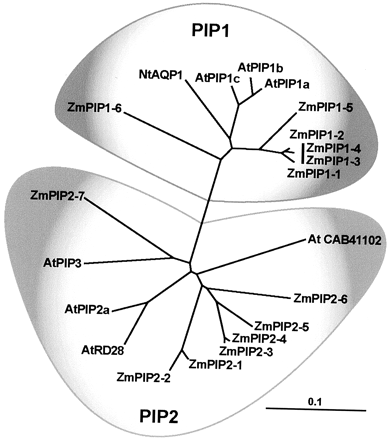 |
Figure
3. Phylogenetic analysis of the maize ZmPIPs and other plant PIPs.
Accession numbers of ZmPIPs are shown in Table I. Other plant PIP
accession numbers are indicated in the tree or (in parentheses): NtAQP1
(AJ001416), AtPIP1c (AAF81320), AtPIP1b (AAC28529), AtPIP1a (CAB71073),
AtRD28 (AAD18141), AtPIP2a (CAB67649), and AtPIP3 (CAA17774). The
distance scale represents the evolutionary distance, expressed in
the number of substitutions per amino acid.
|
The phylogenetic
tree of PIPs indicates that the multiplicity of most maize and Arabidopsis
PIPs must have emerged relatively late during evolution, after the monocot-dicot
divide. Five of the six maize PIP1 sequences cluster in one branch, separate
from all Arabidopsis PIP1 sequences. ZmPIP1-6 forms a third PIP1 branch.
In a similar manner, six of the seven maize PIP2 sequences cluster in one
group. ZmPIP2-7 forms a branch with two Arabidopsis sequences (AtPIP3 and
AAC64216.1), and all other 11 AtPIPs cluster in two groups, apart from the
maize sequences (Fig. 3; for simplicity of representation, only selected
Arabidopsis sequences are shown in the tree).
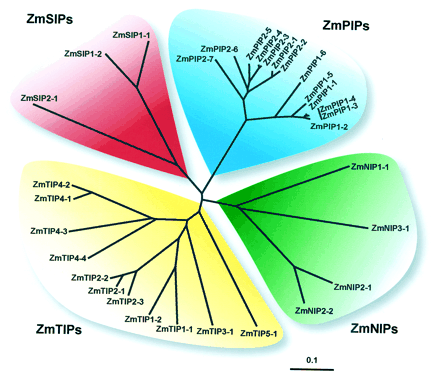
Phylogenetic Analysis of ZmTIPs
The ZmTIP cladogram (Fig. 4) shows that TIPs can be divided into five groups.
TIP1 corresponds to the highly expressed and active -TIPs found in many
plants (Maurel et al., 1993; Chaumont et al., 1998). TIP2 corresponds to
-TIP of Arabidopsis (Daniels et al., 1996). Vacuoles containing -TIP proteins
may act as storage compartments for pigments and vegetative storage proteins
(Jauh et al., 1998). TIP3 corresponds to -TIP, first found in the common
bean (Phaseolus vulgaris) and highly expressed in cotyledons where it is
a component of the membrane that delimits the protein storage vacuole (Johnson
et al., 1990). TIP4 represents a family that also contains NtTIPa, a protein
that transports water and glycerol in X. laevis oocytes (Gerbeau et al.,
1999). A closely related sequence is found in the Arabidopsis database (Fig.
4). The TIP5 group includes a not-yet-characterized Arabidopsis protein
and two proteins from maize and barley (Hordeum vulgare), respectively.
ZmTIP5-1 is characterized by an eight-amino acid residue insertion in the
third loop. In contrast to PIPs, the TIP cladogram (Fig. 4) reveals monocot
and dicot sequences clustered together in each of the five major TIP branches.
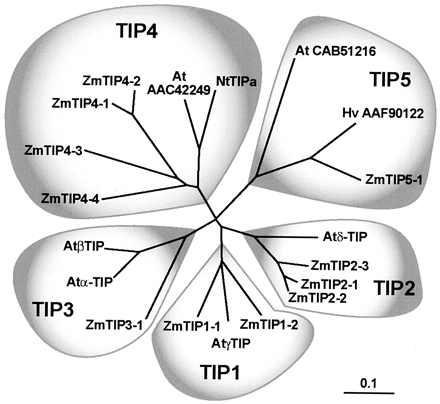 |
Figure
4. Phylogenetic analysis of the maize ZmTIPs and other plant TIPs. Accession
numbers of ZmTIPs are shown in Table I. Other plant TIP accession numbers
are indicated in the tree or (in parentheses): At-TIP (AAF18716), AtTIP
(AAF97261), AtTIP (BAB1264), AtTIP (AAD31569), and NtTIPa (CAB40742).
|
Phylogenetic
Analysis of ZmNIPs
The Nod26-like major intrinsic protein (MIP) subfamily (NIP) is represented
by four members in maize (Fig. 5). One of these (ZmNIP1-1) is most closely
related to four other proteins (GmNod26, LjLIMP2, AtNLM1, and AtNLM2) that
have been found to transport glycerol as well as water (Rivers et al., 1997;
Dean et al., 1997; Guenther and Roberts, 2000; Weig and Jakob, 2000). Two
of the other sequences (ZmNIP2-1 and ZmNIP2-2) are closely related and may
be the result of a more recent duplication. The NIPs and PIPs are the longest
proteins, but NIPs differ mainly from PIPs by the presence of longer C-terminal
tails (eight-30 amino acid residues) that are highly charged in the case
of ZmNIP2-1 (16 out of 41 amino acids) and ZmNIP2-2 (12 out of 35 amino
acids).
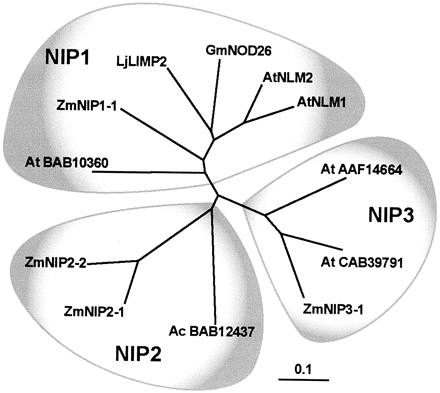 |
Figure
5. Phylogenetic analysis of the maize ZmNIPs and other plant NIPs. Accession
numbers of ZmNIPs are shown in Table I. Other plant NIP accession numbers
are indicated in the tree or (in parentheses): LjLIMP2 (AAF82791), GmNOD26
(AAA02946), AtNLM1 (CAA16760), and AtNLM2 (CAB78893). The distance scale
represents the evolutionary distance, expressed in the number of substitutions
per amino acid.
|
On cladograms
that include water-specific AQPs as well as glycerol transporters from bacteria,
yeast, and mammals, all the sequences cluster into two groups: an AQP cluster
(true AQPs) and a glycerol facilitator-like protein (GLP) cluster (Park
and Saier, 1996; Heymann and Engel, 1999). A similar cladogram that includes
the plant AQPs that transport glycerol, whether in the PIP, TIP, or NIP
cluster, shows that these glycerol transporters are grouped with the AQP
cluster. Froger et al. (1998) identified five conserved amino acid positions
that differ consistently between the two groups. However, in the NIP glycerol
transporters of plants only two out of these five positions follow this
rule (Guenther and Roberts, 2000; Weig and Jakob, 2000). Also, in ZmNIP1-1
and ZmNIP3-1, aromatic (Phe) and aliphatic (Leu/Val) residues are found
in positions P1 and P5, respectively, in accordance with the glycerol transporter
rule identified by Froger et al. (1998). However, ZmNIP2-1 and ZmNIP2-2
have residues that are typical of orthodox AQP. As seen in the cladogram
(Fig. 5), maize NIP1 and NIP3 sequences have counterparts in dicot species.
No dicot orthologs for the NIP2 have been detected in the databases, including
the entire Arabidopsis genomic sequence. However, it is interesting that
a close NIP homolog that clusters in a cladogram to the NIP2 branch sequences
has been reported from the fern Adiantum capillus-veneris (accession no.
BAB12437).
Phylogenetic Analysis of ZmSIPs
The SIPs constitute
a new small subfamily that has been recently identified in Arabidopsis (U.
Johanson and P. Kjellbom, personal communication at the MIP 2000 meeting
in Göteborg, Sweden, July 2000) and in maize by one of us (R. Jung). The
amino acid sequence of this group is the most highly diverged, showing only
16% to 28% identity with the three other groups (Table II). Not only is
there a general divergence over the entire sequence, but there is a striking
lack of conservation in the short helix of loop B, which contains the first
NPA motif (Fig. 6). This motif is represented by Asn-Pro-Thr or Asn-Pro-Leu
in the ZmSIP sequences. In Arabidopsis, the third position can be occupied
by Thr, Cys, or Leu. The second NPA motif is conserved in all maize and
Arabidopsis AQPs. Overall in helix B, there is complete conservation of
6 other amino acids in all the ZmTIPs and ZmPIPs: SGGHXNPAVT. Of these six
positions, only one is conserved in the ZmSIPs. Therefore, it is likely
that ZmSIPs evolved separately from an ancestral gene. The ZmNIPs also show
some divergence in this highly conserved loop (Fig. 6). The SIP cladogram
(Fig. 7) shows that SIPs can be divided into two groups, each of them including
the same number of maize and Arabidopsis proteins.
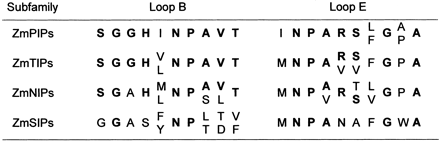 |
Figure
6. Amino acid residues in the NPA motifs of ZmAQPs. The amino acid residues
in the structural loops B and E of each maize subfamily are indicated.
Residues in bold are found in 20 or more of 31 ZmAQPs.
|
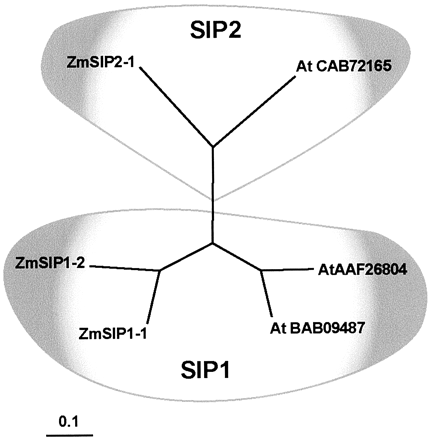 |
Figure
7. Phylogenetic analysis of the maize and Arabidopsis SIPs. Accession
numbers of ZmSIPs are shown in Table I. Arabidopsis accession numbers
are indicated in the tree. The distance scale represents the evolutionary
distance, expressed in the number of substitutions per amino acid.
|
Conserved Motifs
and Amino Acids
The AQP monomers
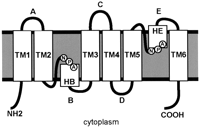
of maize contain 243 to 302 amino acid residues that form two tandem repeats
of three membrane-spanning -helices (TM1-TM6) and with amino and carboxy
termini located on the cytoplasmic side of the membrane. Analysis of the
crystal structures of mammalian AQP1 (Murata et al., 2000) and bacterial
GlpF (Fu et al., 2000) shows that portions of two loops (loop B and loop
E) form -helices that dip halfway into the membrane from opposite sides
and form the aqueous pore. Figure 8 shows a model of ZmPIP1-2 that is based
on the structure of AQP1 (Mitsuoka et al., 1999; Murata et al., 2000) and
on an analysis of putative transmembrane domains based on a hydrophobicity
plot. The amino acids that are conserved in 20 or more of the 31 maize AQPs
are indicated in color. The highest degree of conservation is in the transmembrane
domains and more particularly in the two loops that form the aqueous pore
and contain the two NPA motifs.
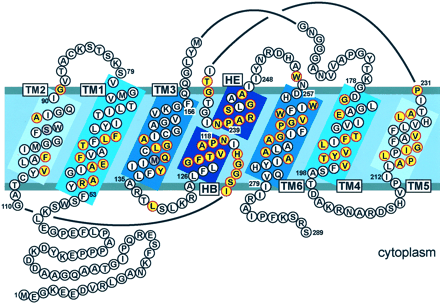 |
Figure
8. A topological model of the maize ZmPIP1-2. The representation is
based on the human AQP1 and bacterial GlpF structures (Fu et al., 2000;
Murata et al., 2000), and shows the six transmembrane helices (TM1-TM6)
and the two short helices in the structural loops B and E (HB and HE).
Residues in yellow with thick red circles are highly conserved among
ZmAQPs (found in 20 or more of the 31 ZmAQPs). Residues in pink indicate
the position of a highly conserved residue present in 20 or more of
the 31 ZmAQPs but absent from ZmPIP1-2 (97Ser and 140Met of ZmPIP1-2
are replaced by an Ala).
|
A number of
amino acid positions are conserved in the two tandem repeats of the protein.
These include Glu 60 and 184 in TM1 and TM4, Thr 64 and 188 in TM1 and TM4,
Gly 145 and 264 in TM3 and TM6, and Gly 113 and 234 in loops B and E, respectively.
In addition to these residues, a large number of amino acid positions are
conserved either in the first half or the second half of the protein. Many
of the positions conserved in maize AQPs are also conserved in other AQPs
(Heymann and Engel, 2000; Murata et al., 2000). Maize has some conserved
positions not conserved in other AQPs; For example, Arg 55 and Ala 56 in
TM1 are highly conserved, as are Ala 94 and Phe 102 in TM2; Leu 144 in TM3;
Phe 189 in TM4; and Ala 214, Leu 216, and Leu 227 in TM5.
In mammalian
AQP1, the positions of the two functional loops that form the aqueous pore
are stabilized through ion pairs and hydrogen bonds of highly conserved
amino acids. These are also conserved in the maize AQPs and on the basis
of the structure of AQP1 (Mitsuoka et al., 1999, 2000) we can predict the
following interactions. His 115 in loop B forms an ion pair with Glu 60
in TM1 and Arg 241 in HE is connected by a salt bridge to Glu 184 in TM4.
Ser 112 in loop B forms a hydrogen bond with Tyr 137 (TM3), further stabilizing
loop B.
Expression of Maize AQP Genes
Because we
used so many different libraries prepared at different times from plants
grown under slightly different conditions, the number of times a specific
cDNA appears gives only a rough estimate of its abundance in the mRNA population.
Most abundantly expressed are ZmTIP1-1 (a -TIP-like sequence), ZmTIP2-1,
most of the members of the ZmPIP1 family, and ZmPIP2-1 (Table III). Ten
of the sequences were found only a few times (between one-10 times) and
another eight were found less than 20 times. These include all the NIPs
and SIPs, which were also the last groups of AQPs to be identified in plants.
Most of the more abundantly expressed sequences were found in the various
plant organs that were examined. The tissue distribution of the 124 ESTs
of ZmPIP1-3 and ZmPIP1-4 is shown in combined numbers (Table III). Because
of their very close relationship (approximately 98% identity of the nucleotide
sequence), only one single cluster was formed by the ESTs of these two PIPs.
Using computer algorithms, it was not possible to faithfully deconvolute
this cluster. However, by visual inspection of a representative sample set
of high-quality ESTs and by considering signature sequences, we can conclude
that about 25% of the ESTs appear to represent ZmPIP1-3 and 75% represent
ZmPIP1-4. The distribution of both cDNAs did not appear to differ significantly.
Table III.
Distribution of cDNAs in maize libraries
|
|
|
|
| Name |
TCa |
RTb |
Root |
Vc |
Sum |
|
| ZmPIP1-1 |
17 |
65 |
38 |
78 |
198 |
| ZmPIP1-2 |
3 |
45 |
9 |
26 |
83 |
| ZmPIP1-3 |
8 |
66 |
12 |
38 |
124 |
| ZmPIP1-4 |
| ZmPIP1-5 |
1 |
0 |
18 |
6 |
25 |
| ZmPIP1-6 |
0 |
0 |
0 |
1 |
1 |
| ZmPIP2-1 |
29 |
99 |
17 |
56 |
201 |
| ZmPIP2-2 |
2 |
24 |
9 |
8 |
43 |
| ZmPIP2-3 |
1 |
11 |
8 |
9 |
29 |
| ZmPIP2-4 |
2 |
0 |
27 |
9 |
38 |
| ZmPIP2-5 |
1 |
2 |
8 |
6 |
17 |
| ZmPIP2-6 |
12 |
6 |
9 |
12 |
39 |
| ZmPIP2-7 |
0 |
0 |
0 |
3 |
3 |
| ZmTIP1-1 |
29 |
114 |
27 |
66 |
236 |
| ZmTIP1-2 |
0 |
6 |
0 |
3 |
9 |
| ZmTIP2-1 |
5 |
29 |
56 |
27 |
117 |
| ZmTIP2-2 |
0 |
0 |
27 |
1 |
28 |
| ZmTIP2-3 |
2 |
0 |
11 |
0 |
13 |
| ZmTIP3-1 |
9 |
6 |
0 |
0 |
15 |
| ZmTIP4-1 |
0 |
0 |
3 |
8 |
11 |
| ZmTIP4-2 |
0 |
3 |
0 |
8 |
11 |
| ZmTIP4-3 |
0 |
6 |
0 |
0 |
6 |
| ZmTIP4-4 |
0 |
5 |
0 |
0 |
5 |
| ZmTIP5-1 |
0 |
1 |
0 |
0 |
1 |
| ZmNIP1-1 |
0 |
14 |
0 |
3 |
17 |
| ZmNIP2-1 |
0 |
0 |
0 |
3 |
3 |
| ZmNIP2-2 |
0 |
0 |
0 |
1 |
1 |
| ZmNIP3-1 |
2 |
12 |
3 |
0 |
17 |
| ZmSIP1-1 |
1 |
6 |
0 |
0 |
7 |
| ZmSIP1-2 |
3 |
12 |
0 |
2 |
17 |
| ZmSIP2-1 |
0 |
4 |
0 |
0 |
4 |
|
| Total |
127 |
536 |
282 |
374 |
1,319 |
|
|
a Tissue culture. b Reproductive tissue. c Aereal vegetative tissue.
|
It is clear
that a more detailed analysis is needed to determine the tissue and cell
type specific expression of the individual genes. The information given
in Table III provides a sound basis from which to proceed with such an analysis.
DISCUSSION
By screening
a very large database of maize ESTs we identified a number of AQP genes
and for 31 of these we were able to obtain complete nucleotide sequences.
This large number is not surprising because Arabidopsis contains 35 AQP
genes (Weig et al., 1997; Kjellbom et al., 1999, and our own analysis of
the genome). AQP cDNAs are often very difficult to clone in Escherichia
coli and we expect that some additional maize AQPs will be forthcoming.
We obtained four partial sequences (one PIP, two TIPs, and one NIP) for
which we were unable, after repeated attempts, to obtain full-length sequences.
This is probably caused by the hydrophobicity of the proteins, which could
disrupt bacterial membranes and impair bacterial growth, even if they are
expressed at a very low level. If we obtain these full sequences they will
be submitted to GenBank as we obtain them.
Maize AQPs Form Four Subfamilies
The maize AQP
sequences can be grouped into four subfamilies, which we named PIPs, TIPS,
NIPs, and SIPs. This nomenclature preserves the earlier names of TIPS and
PIPs, which denote sequence similarity rather than subcellular location.
We do not know if all the PIPs and TIPs are located in plasma membranes
and tonoplasts, respectively. The name NIP refers to the Nod26-like MIPs
previously called Nod-like MIP (NLM; Weig et al., 1997; Weig and Jakob,
2000). The name SIP refers to small basic integral proteins, identified
in maize by one of us (R. Jung) and in Arabidopsis (by U. Johanson in an
oral communication at the MIP2000 meeting in Gothenburg, Sweden in July
2000). These proteins are not smaller than TIPs but appear to be more basic
and as a group have a slightly higher pI than the TIPs.
The distribution
of sequences between the four major subfamilies is very similar in maize
and Arabidopsis, where 13 PIPs, 10 TIPs, nine NIPs, and three SIPs can be
found in the recently completed sequence of the entire genome. In maize,
if we count full-length and partial sequences, we have 14 PIPs, 13 TIPs,
five NIPs, and three SIPs. Unlike the Arabidopsis genome, the maize genome
has not yet been sequenced. The broad representation of AQPs in the four
categories gives us confidence that we have obtained most of the maize AQPs.
Moreover, AQPs from maize and Arabidopsis are found in each group of the
different subfamilies, except for the NIP2 group where no Arabidopsis counterpart
has been detected in its genome. Overall, this observation suggests that
the separation into the different groups had occurred before the monocot-dicot
divergence and that the ancestral gene of these groups encoded a protein
with a specific biological role. The persistence of these groups in monocots
and dicots is also an indication of the crucial role of AQPs in water and
solute relations in plants.
The phylogenetic
analysis of the four subfamilies showed that, in many cases, the proteins
in a given species are found on a single branch (without members from another
species), indicating a number of recent DNA duplication events arising after
the monocot-dicot separation. As already outlined above, this is particularly
striking in the PIP tree, where five ZmPIP1 and six ZmPIP2 proteins are
found on the same branch in each respective group. The same phenomenon is
much less pronounced in the TIP cladogram, where most subgroups contain
monocot as well as dicot sequences. This independent, and apparently evolutionary,
late emergence of novel PIP genes could indicate similar adaptive advantages
that were gained in the two large angiosperm clades, probably in response
to similar selective pressures.
This leads
to the crucial question: Why are there so many different AQPs in a single
plant species? The separation of AQPs in different subfamilies and groups
may reflect a specialization of the function and the localization. The water
channel activity of only four maize AQPs has been tested in X. laevis oocytes,
but these data and those obtained in other species indicate a differential
regulation of the activity. ZmTIP1-1 is the most highly expressed tonoplast
AQP, corresponding to the Arabidopsis -TIP (Barrieu et al., 1998; Chaumont
et al., 1998). Members of the TIP4 group may transport solutes in addition
to water as demonstrated for NtTIPa, which transports urea and glycerol
in addition to water (Gerbeau et al., 1999). As is the case for the other
plasma membrane PIP2 proteins tested so far, ZmPIP2-5 is a good water channel,
but ZmPIP1-1, ZmPIP1-2, and other PIP1 homologs show a poor water-transport
activity in oocytes (Chaumont et al., 2000). NIP proteins have been found
to transport glycerol as well as water (Dean et al., 1997; Rivers et al.,
1997; Guenther and Roberts, 2000; Weig and Jakob, 2000).
More recent
duplication events giving rise to close isoforms in a single species could
be a way to control specific expression according to developmental and environmental
conditions. For instance, ZmPIP2-2 is mostly expressed in reproductive tissue
and ZmPIP2-4 in roots. This could also be true for proteins present in a
specific group, such as the TIP3 members, which are found in seed and cotyledons
(At-TIP).
ZmPIP1-3 and
ZmPIP1-4 obviously are also the result of a very recent gene duplication.
Both genes encode identical proteins and their transcripts show a similar
tissue distribution. The result of having two genes may be an increased
expression level due to a gene dosage effect of the duplicated genes. A
recent study (Lynch and Conery, 2000) based on genome-wide analyses of different
organisms discusses frequent gene duplications and their importance for
the evolution of a species and its evolutionary fate. Because of its large
size, the AQP gene family in plants is well suited to test such a phylogenetic
hypothesis.
It is interesting
that we obtained an aberrant SIP cDNA that also contained exonic sequences
of an unrelated gene. This cDNA is not a cloning artifact, but rather it
is derived from a pseudogene transcript. Several clones of this cDNA were
isolated from independently constructed libraries, all originating from
a cultured cell line of the maize cv Black Mexican Sweet. The derived amino
acid sequence of this pseudogene contains several stop codons and was not
included in the phylogenetic analysis.
Structural Features of Water and Glycerol Transporters
Mammalian AQP1 has recently been crystallized and its three-dimensional
structure determined at 3.8-Å resolution (Mitsuoka et al., 1999; Murata
et al., 2000). We modeled ZmPIP1-2 based on the structure of AQP1 (Fig.
8). Sequence analysis revealed the conservation of many amino acid positions
(about 60), especially in the two loops that create the aqueous pore. In
addition, amino acid residues in different domains that stabilize the structure
of the pore through a salt bridge, an ion pair, and a hydrogen bond are
completely conserved in all maize AQPs. Determination of the structure of
a plant AQP (common bean -TIP) at much lower resolution (7.7 Å) has indicated
that this plant AQP has the same general structure as AQP1 (Daniels et al.,
1999).
Some AQPs are
water specific; others, such as GlpF from E. coli, are glycerol specific,
and yet others have a mixed function. Several AQPs that transport glycerol
have been identified in plants (Rivers et al., 1997; Biela et al., 1999;
Dean et al., 1999; Gerbeau et al., 2000; Guenther and Roberts, 2000; Weig
and Jakob, 2000), although it is not known that they transport glycerol
in planta. On a comprehensive cladogram that includes all AQPs, these glycerol
transporters are grouped with the water-transporting AQPs and not with the
glycerol transporters of prokaryotes and the mixed-function AQPs of mammals
(Heymann and Engel, 1999).
Froger et al.
(1998) identified five amino acid positions that appear to be highly conserved
in the glycerol transporters, but only two of these are conserved in the
glycerol transporters of plants (see also Weig and Jakob, 2000). Also in
maize, only two of these positions are conserved in ZmNIP1-1 and ZmNIP3-1,
which are the putative glycerol transporters, but are not conserved in ZmNIP2-1
and 2-2. The structural analysis of GlpF (Fu et al., 2000) does not indicate
why these conserved residues might be important. Fu et al. (2000) identified
the residues that interact with glycerol in the channel as well as the hydrophobic
residues that line the amphipathic channel, and there is better conservation
of these between GlpF and maize NIPs. Of seven channel residues that interact
with glycerol via hydrogen bonds, five are conserved. There is a substitution
of Phe200 (GlpF numbering) with Gly or Ala in ZmNIPs and of Ala201 with
Ser. However, it is the carbonyl groups of these residues that are important
for hydrogen bonding to glycerol rather than their functional groups. The
carbonyls are oriented by hydrogen bonding of backbone NH to a highly conserved
Glu in TM4. Future functional analysis will reveal whether the maize NIPs
are glycerol channels.
MATERIALS AND
METHDS
cDNA Libraries and EST Databases
In toto, 215
maize (Zea mays) cDNA libraries were constructed covering all major maize
tissue types, including different developmental stages of these tissues,
tissues from plants under biotic and abiotic stress conditions, tissues
and cell cultures responding to chemical treatments, and tissues isolated
from mutant maize lines. RNA was isolated from maize tissues using TriZol
Reagent (Gibco-BRL, Gaithersburg, MD). cDNA synthesis was performed using
the SuperScript II kit and cloned into NotI/SalI sites of the pSPORT1 vector
(Gibco-BRL) or by using the cDNA Synthesis Kit (Stratagene, La Jolla, CA)
and cloning into XhoI/EcoRI sites of the pBluescript SK+ vector (Stratagene).
Inserts of randomly picked clones were sequenced by Human Genome Systems
(Rockville, MD) or at the DuPont genomics facility (Newark, DE) from the
5' end to obtain ESTs. To a limited amount clones were picked after library
normalization or from subtracted libraries. The sequence information of
472,890 maize EST accessions is maintained in the central DuPont/Pioneer
Hi-Bred genomics database and is accessible via database interface software.
Clustering, Identification, and Analysis of Maize AQP EST and cDNA Sequences
EST sequences
were first evaluated using PHRED-assigned quality scores (Ewing et al.,
1998) and, after removal of short, low-complexity, and low-quality sequences,
the similarity relationship (clustering) between ESTs was established using
the BLAST algorithm (Altschul et al., 1990). EST clusters were subsequently
subjected to a sequence assembly process using the PHRAP algorithm (http://www.phrap.org/phrap.docs/phrap.html)
and about 38,000 contigs and 92,000 singletons were made. The resulting
database of contigs and singletons was systematically searched (BLAST) for
AQP-related sequences using publicly available AQPs from Escherichia coli,
yeast (Saccharomyces cerevisiae), plants, and animals. As a result, about
1,300 EST accessions formed an initial group of about 200 AQP-related sequences,
singletons, and contigs. These sequences were thoroughly inspected for over-
and under-clustering, visually and by pair-wise sequence alignments (BLAST,
ClustalW, Gap), which resulted in 84 putatively unique sequences. The longest
clone representing each of these sequences was obtained from the libraries
and both strands of each insert were sequenced either by primer walking
or by sequencing of nested sets of deletion sub-clones after transposition.
The identification and clustering of unique maize AQPs was further refined
by repeated pair-wise BLAST searches and by searches of the entire EST database
using both the obtained full-length insert nucleotide sequences and their
deduced encoded amino acid sequences. After several rounds of reiterative
analysis and sequencing, 36 complete nucleotide sequences of unique maize
AQPs, 32 encoding full-length cDNA, and four encoding partial cDNA were
identified in the current DuPont/Pioneer Hi-Bred maize genome database.
An analysis of maize ESTs in the public databases did not turn up additional
unique AQP sequences.
The transcript
expression profile of each unique maize AQP was estimated by tallying the
tissue distribution of clustering ESTs.
The sequence
alignments were performed by CLUSTAL X (1.8; Thompson et al., 1997). The
pair-wise alignments were calculated by the dynamic programming method.
The multiple alignments used the series of GONNET matrices. Trees were calculated
using the Neighbor-Joining method and displayed using TreeView (Page, 1996).
We used the gene product names when available and the systematic open reading
frame names otherwise. Figures were prepared in Illustrator 9.0 (Adobe Systems
Incorporated, San Jose, CA).
|